What is BRASS?
BRASS is a family of in silico pathogenicity predictors specific for BRCA1/2 variants. We have two predictors per protein, MLR and NN. For a given variant, MLR provides an estimate of its HDR activity, while NN provides a binary (neutral/pathogenic) estimate of its impact on the carrier. BRASS is from the Clinical and Translational Bioinformatics research group at Vall d'Hebron Institute of Research.
How can I analyze my variant?
You can submit your variant in the query page of BRCA1 or BRCA2 protein, indicating the predictor you prefer (MLR or NN), and the native amino acid, residue and mutated amino acid of the missense variant. Afterwards, you will be redirect to the prediction page.
Why is my variant not accepted?
We map your variant to the canonical isoform of the protein provided by UniProt (UniProt Consortium, 2023). If the native amino acid of your variant does not match the native amino acid in the canonical isoform, the program will raise an error message. This may happen if your sequence is based on another isoform of UniProt, NCBI or Ensembl.
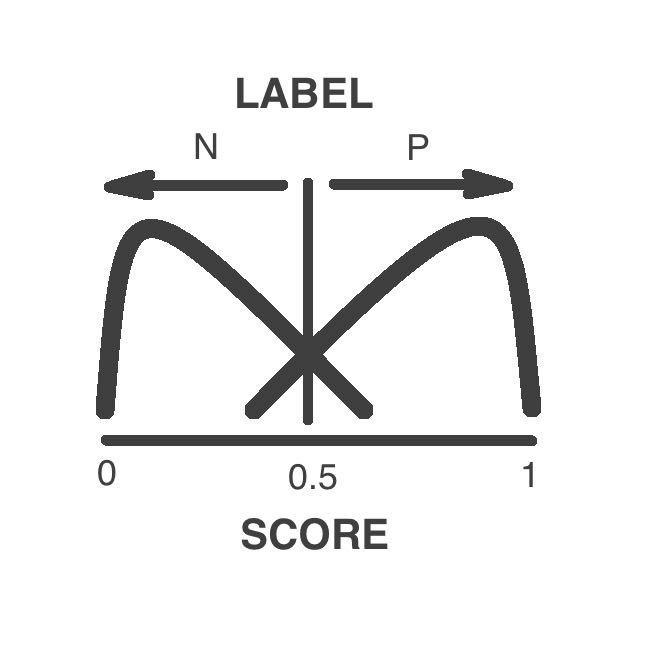
What is the output for the pathogenicity predictor NN?
We provide:
Label: a binary output describing the estimated phenotype of the variant's carrier: it can either be neutral or pathogenic.
Score: it is an estimate of the probability of the pathogenicity of the variant. It is used to generate the discrete prediction (neutral: score <0.5; pathogenic: score >=0.5).
Reliability: a measure of how accurate is the prediction. It varies between 0 (highly unreliable) and 10 (highly reliable). It is graphically represented using a combination of filled and empty circles: the more filled circles, the more reliable the prediction is.
What is the output for the functional predictor MLR?
We provide:
Label: a binary output indicating whether the variant is expected or not to disrupt the normal HDR function of the protein.
Score: an estimate of the output of the HDR assay for the variant, according to Farrugia et al.
How was the pathogenicity predictor NN derived?
NN is a protein-specific predictor trained following the protocol described in Riera et al. (Human Mutation, 2016):
We obtained a set of manually curated BRCA1/2 variants.
We used these variants to train a neural network model.
We estimated the prediction performance using a leave-one-out cross-validation procedure.
How was the functional predictor MLR obtained?
MLR predicts the impact of a variant on the HDR activity of BRCA1/2 proteins. It was obtained using the following protocol:
Collect a set of variants with known HDR values from the work of Starita et al. for BRCA1, and Farrugia et al. for BRCA2.
Fit a multiple linear regression model to these values, separately for each protein. The independent variables of the model where two conservation-based measures from our earlier work (Riera et al. (Human Mutation, 2016)) and the value of the Blosum62 matrix (Henikoff et al. (PNAS, 1992)) for the amino acid replacement
Estimate the model performance by cross-validation leave-one-out cross-validation procedure.
Can I download all the predictions for my protein?
Yes, you can download all the pre-calculated predictions here. The file is in csv format and has the following columns:
| # | Field | Description |
|---|---|---|
| 1 | Gene | HGNC official gene symbol |
| 2 | Protein | Uniprot accession number |
| 3 | Variant | Missense variant from the canonical isoform |
| 4 | Prediction | Predicted functional consequence of the variant |
| 5 | Score | Numerical score of the pathogenic prediction |
Is there any additional information provided with the pathogenicity prediction?
Yes, together with the prediction we provide information that may help extending/complete our understanding of the variant's impact. We also give a series of details to help understand the origin of the prediction. This information is structured in three broad sections:
Top of the page. We find our prediction and a series of links to predictions of the variant from different tools.
Section "Interpreting the variant". Here, we find a series of links to different resources where we can access information on existing clinical evidence, on the biological relevance and frequency of the variant, on the protein and the gene, etc. In this section, a dynamic graph will allow us to locate the variant in the sequence of the protein and put this location in the context of the known protein domains, gene exons, functionally relevant residues and the pathogenic and neutral variants used to train the BRASS predictor.
Section "Understanding the prediction". Here, we find a series of plots where we can see what is the value of our variant for the different properties used in the pathogenicity prediction. In these plots we also see the value of these properties for the variants of the training set. Analysis of these plots indicates how much our variant resembles pathogenic or neutral variants (from the training dataset), and helps us understand which property, or properties, where at the origin of the prediction.