What is ProteinSpecific predictor?
ProteinSpecific predictor is a tool to predict the functional significance of missense variants of more than 80 proteins involved in diseases with an inheritance component from the Clinical and Translational Bioinformatics research group at Vall d'Hebron Institute of Research.
How can I analyze my variant?
You can submit your variant in our query page indicating the native amino acid, the residue and the mutated amino acid. Afterwards, you will be redirect to the prediction page.
Why is my variant not accepted?
We map your variant to the canonical isoform of the protein provided by UniProt (UniProt Consortium, 2023). If the native amino acid of your variant does not match the native amino acid in the canonical isoform, the program will raise an error message. This may happen if your sequence is based on another isoform of UniProt, NCBI or Ensembl.
What information we provide for a pathogenicity prediction?
We provide:
Label: the variant is classified as pathogenic or neutral according to its functional consequence.
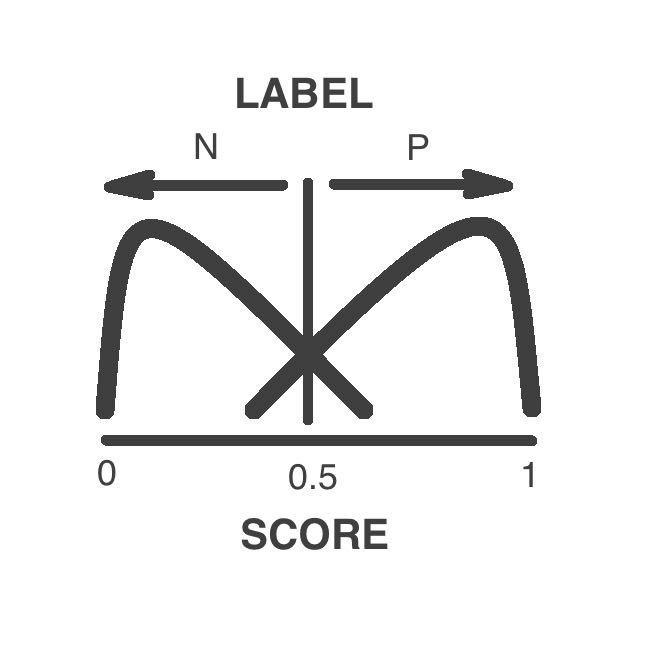
Score: the numerical score of the functional consequence of the variant. It has a continuous scale from 0 to 1, being 0 a neutral and 1 a pathogenic variant. The threshold between pathogenic and neutral variant is at 0.5.
Reliability: measures the accuracy of the prediction. It has a continuous scale from 0 to 1, being 1 a truthful prediction.
How are the pathogenicity predictions calculated?
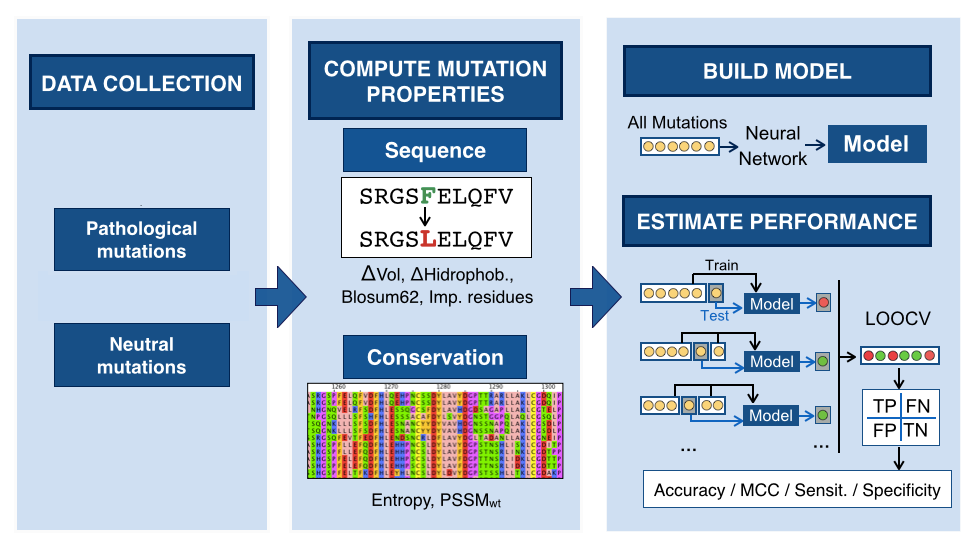
These predictions are calculated by a machine learning algorithm previously trained with a set of already known variants from the literature. To develop the predictor, we followed these steps:
Collect the pathogenic and neutral variants of the proteins
Decipher the features able to discriminate between pathogenic and neutral variants
Build the model by training the machine learning algorithm with the set of features of the known variants
Estimate the model performance by cross-validation to ensure the reliability of the predictor
Riera et alt., Human Mutation, 2016
What is the performance of ProteinSpecific predictor?
The ProteinSpecific predictor has been evaluated and compared to the state of the art predictors. The Matthews Correlation Coefficient (MCC) per gene and predictor is:
In mean, the performance metrics per predictor are:
| Sensitivity | Specificity | Accuracy | MCC | Coverage | |
|---|---|---|---|---|---|
| ProteinSpecific | 0.881 | 0.866 | 0.873 | 0.703 | 100% |
| PON-P2 | 0.917 | 0.802 | 0.879 | 0.724 | 51% |
| PolyPhen-2 | 0.905 | 0.791 | 0.834 | 0.649 | 98% |
| CADD | 0.953 | 0.655 | 0.766 | 0.588 | 27% |
Can I download all the predictions for my protein?
Yes, you can download all the pre-calculated predictions here. The file is in csv format and has the following columns:
| # | Field | Description |
|---|---|---|
| 1 | Gene | HGNC official gene symbol |
| 2 | Protein | Uniprot accession number |
| 3 | Variant | Missense variant from the canonical isoform |
| 4 | Prediction | Predicted functional consequence of the variant |
| 5 | Score | Numerical score of the pathogenic prediction |
Is there any additional information provided with the pathogenicity prediction?
The results report a great amount of information related to the variant divided in different sections:
Prediction: prediction of the functional consequence of the variant along with its score and reliability.
Other Predictors: functional consequence of the variant predicted by other standard tools such as PON-P2, PolyPhen-2, SIFT and CADD predictors.
Variant Annotation: known the clinical evidence, biological relevance, population allele frequency and other information about your variant from several databases such as ClinVar, UniProt, dbSNP and ExAC.
Biomedical Information: links to several resources about the disease (DECIPHER, HPO, GeneReview, Malacards, MedGen, OMIM and Orphanet databases), the protein (UniProt database), the tridimensional structure (PDB database), the protein-protein interactions (STRING database), the metabolic pathways (REACTOME database), and the gene (Ensembl, GeneCards, HGNC and NCBI databases).
Protein Plot: distribution of several features along the protein such as known pathogenic and neutral variants, biological relevant residues, functional domains and gene exons.
Predicted functional consequence: localization of the score of the variant in the distribution of scores of known pathogenic and neutral variants.
Explanatory variables of the prediction: localization of the features of the variant in the distribution of features of known pathogenic and neutral variants.